Case Study 21
The patient in this case study was a male diagnosed with infantile nephrotic cystinosis at the age of eleven months. Nephrotic cystinosis is a rare autosomal recessive disorder resulting in the accumulation of cystine crystals primarily in the kidney as well as other organs, including the bone marrow. These crystals are almost never seen in the peripheral blood. Cystinosis presents between six and twelve months of age. The clinical characteristics include failure to thrive, progressive renal failure, loss of muscle function, photophobia and fair hair. Diagnosis is made by measuring the level of cystine in peripheral blood white cells.
By the age of eleven years, the patient’s condition had progressed to a stage which involved renal failure requiring peritoneal dialysis and frequent blood transfusions. At the age of thirteen, he received a renal transplant, the kidney being donated by a family member. He was then started on a regimen of immunosuppressive therapy.
By the age of twenty the patient was experiencing neurological abnormalities. He had difficulty in walking and swallowing, loss of memory and progressive loss of speech as well as diminished intellectual function. He continued to receive immunosuppressive therapy for his renal transplant.
At the age of twenty-six he presented to the
Casualty Department. He had cervical and mediastinal
lymphadenopathy and splenomegaly. A full blood count
was performed with the following results:
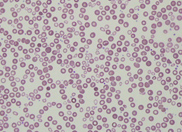
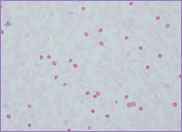
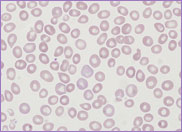
Hb 115 g/L, WBC 5.4 x 109/L and platelet
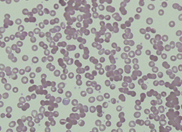
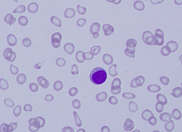
count 197 x 109/L. The peripheral blood
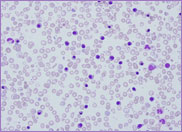
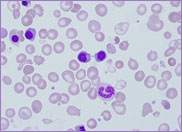
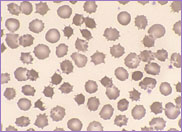
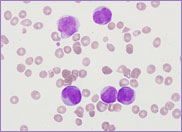
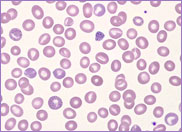
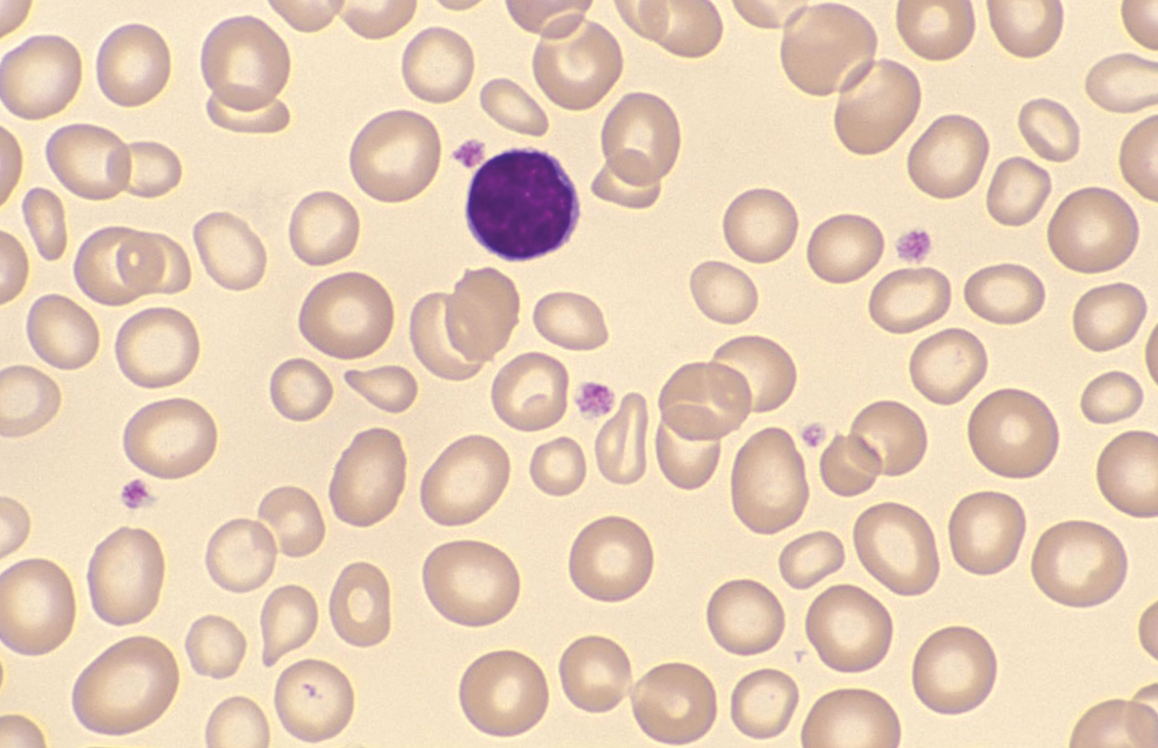
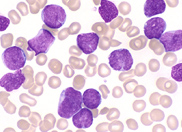
film revealed the presence of circulating blast
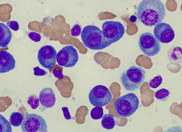
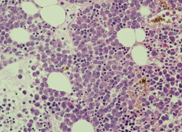
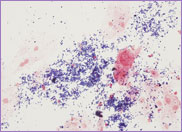
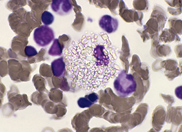
cells. A bone marrow biopsy was performed. The bone
marrow was hypercellular with 70% blasts. The blasts
had a high N/C ratio; the nuclei were variable from
round to irregular and folded in shape. They
contained one to four distinct nucleoli.
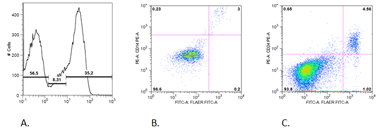
Immunophenotyping was performed with the following
results:
CD45+, HLA-DR–,
CD19+, cytoCD79a+,
cytoCD3+, CD2+,
CD5+, CD7+, CD38+
and CD117+.
CD4–, CD8–.
A diagnosis of T-cell lymphoblastic lymphoma/leukaemia was made.
CD2, 5 and 7 are the most commonly expressed markers
in T-cell lymphoma/leukaemia. CD3 is often present
in the cytoplasm but not on the surface. CD4 and CD8
may be expressed or may be negative.
Co-expression of CD79a and CD117 has been described on primitive T cell lineages however the expression of CD19, normally a B cell marker, is unusual.
A cytogenetic analysis on the available cells revealed a 46, XY karyotype. Subsequent fluorescence in situ hybridisation (FISH) studies showed no evidence of BCR-ABL 1 gene rearrangements.
Post-Transplant Lymphoproliferative Disease
Post-Transplant Lymphoproliferative Disorders (PTLD) are a well-known complication of immunosuppressive treatment after solid organ transplantation. Any lymphoma/leukaemia that occurs in the post-transplant population is considered to be a type of PTLD. The median time from transplant to diagnosis is fifteen years. The majority of post transplant lymphoma/leukaemias are B cell in origin and are associated with EBV infection. T-cell variants are very rare. The patient in this study was EBV positive.
The risk factors of PTLD are intensity of immunosuppression, type of organ transplanted, Epstein-Barr virus (EBV) seronegativity and the duration of the immunosuppression. EBV seronegativity pre-transplant is a powerful predisposing factor for the development of PTLD. Studies show that EBV seronegative patients experience a 10-76 fold greater incidence of PTLD when compared to seropositive counterparts.
The patient in this study died suddenly, two months post chemotherapy induction.