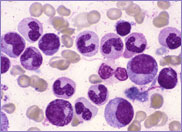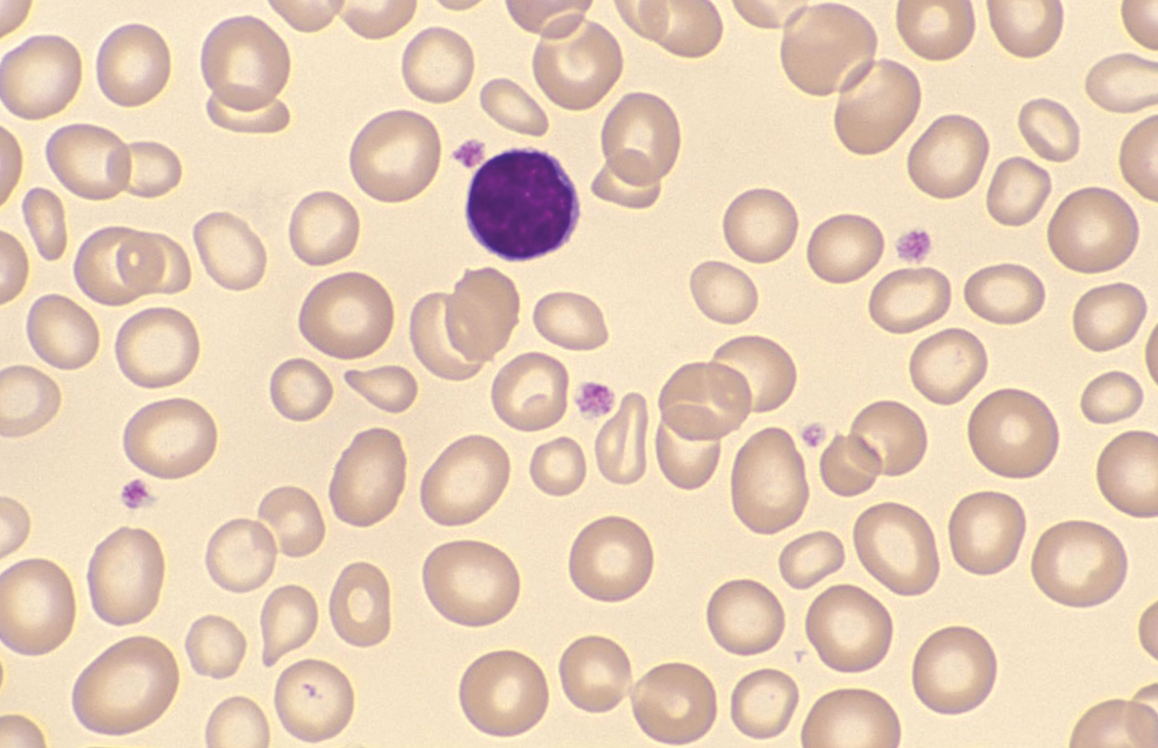
Case Study 1
The blood film of a patient with either hereditary stomatocytosis or Southeast Asian ovalocytosis will show stomatocytes as a common feature. However, there are important differences in the red cell parameters that permit one to differentiate morphologically between the two disorders.
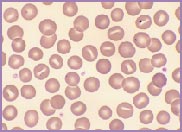
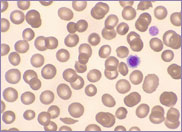
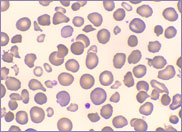
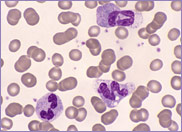
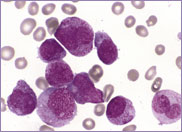
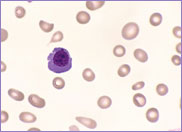
Macrocytic stomatocytes as well as post-splenectomy changes, in a case of hereditary stomatocytosis (hydrocytosis).
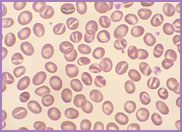
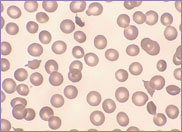
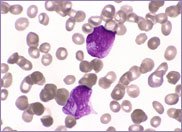
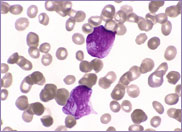
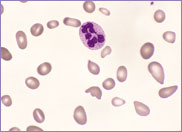
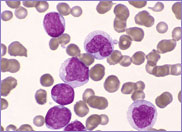
Oval-shaped stomatocytes, some with two transverse slits, in a case of Southeast Asian ovalocytosis.
Hereditary stomatocytosis, often referred to as hereditary hydrocytosis, is a very rare autosomal dominant disorder, characterized by a defect in the Na+ / K+ exchange pump. There is an imbalance between the amount of Na+ entering the red cell and the amount of K+ exiting the cell. The red cell swells as it gains cations and water, transforming from a discocyte to a stomatocyte. These swollen cells are osmotically fragile. They become rigid, requiring extremely large amounts of energy to protect them against lysis.
The red cells of hereditary stomatocytosis lack stomatin (protein 7.2b), an integral red cell membrane protein. The exact function of stomatin is unknown, but it is clearly important in regulating membrane sodium permeability.
The laboratory findings in hereditary stomatocytosis include a macrocytic anaemia. The blood film shows stomatocytes and round macrocytes. The mean cell volume (MCV) is characteristically elevated in the range of 110-150 fL, and there is a correspondingly low mean corpuscular haemoglobin concentration (MCHC) in the range of 240-300 g/L. An elevated red cell Na+ concentration and a reduced K+ concentration, as well as an increased total monovalent cation content, are noted. These abnormal red cells are sequestrated on passing through the spleen, giving rise to extravascular haemolysis. The haemolytic anaemia is severe with a reticulocytosis of >10%. Splenectomy has proven to be the most beneficial treatment in reducing the degree of haemolysis.
Southeast Asian ovalocytosis is also an autosomal dominant disorder. It is very prevalent, occurring in up to 30% of aboriginal people from New Guinea, Malaysia, and Melanesia.
The red cells of Southeast Asian ovalocytosis have a characteristic appearance on the blood film. They are often described as stomatocytic elliptocytes. Instead of being discocytes, they have a slit-like area of central pallor. A small proportion of these stomatocytes have two transverse slits, giving the appearance of double stomatocytes. The red cell membrane of these stomatocytes is very rigid. This abnormality results from increased ankyrin binding and decreased protein 3 mobility, leading to the production of rigid red cells. This rigidity is a protective mechanism against all strains of malaria. There is no abnormality in the red cell Na+/K+ exchange pump.
The laboratory features of Southeast Asian ovalocytosis are unremarkable. The stomatocytes have a normal lifespan; hence, there is no anaemia. The MCV and MCHC are both characteristically within the normal range. In fact, nowadays, with the use of cell analyzers to auto-validate results without examining blood films in the presence of normal blood cell parameters, laboratories diagnose fewer cases of this asymptomatic condition.